
EXCESS MENTAL DISORDERS
https://acamh.onlinelibrary.wiley.com/doi/abs/10.1111/jcpp.14072
https://usatoday.com/lead-gas-worsened-mental-health-gen-x
Leaded gas created a mental health crisis for this generation
by Ken Alltucker┬Ā /┬Ā December 4, 2024
“Researchers have long warned about the harmful effects of lead in paint, pipes and other products. But another once-widespread source ŌłÆ leaded gasoline ŌłÆ might have harmed the mental health of a generation. Gen X bears an extra burden of conditions such as depression, anxiety, ADHD and neurotic behavior because of the leaded gasoline they were exposed to as children, according to a┬Āstudy published Wednesday in the peer-reviewed Journal of Child Psychology and Psychiatry. Leaded gas was banned in the United States in 1996, but the study said years of exposure during development made them particularly vulnerable. Lead gas peaked from the mid-1960s through the mid-1970s, and children born during that era would later develop some of the highest rates of mental health symptoms, the study said. The study also linked leaded gas to “disadvantageous” traits, such as struggling to concentrate, stay on task or organizing thoughts.
“I tend to think of Generation X as ‘generation lead,'” said Aaron Reuben, a study co-author and assistant professor of clinical neuropsychology at the University of Virginia. “We know they were exposed to it more and we’re estimating they have gone on to have higher rates of internalizing conditions like anxiety, depression and symptoms of attention deficit hyperactivity disorder.” Reuben and researchers from Florida State University examined health survey and national gas use data to estimate the amount of lead in people from 1940 through 2015. They used that data to estimate the mental health effects of such exposures. Researchers linked the lead exposure to an estimated 151 million “excess mental disorders” in the United States over the 75-year period.
The estimates should be “considered a floor” because it relies mainly on gas and not exposure from lead in paint and pipes, Reuben said. Although lead paint was banned in 1978, it still can be found in older homes and present hazards to children who ingest paint chips, for example. The Environmental Protection Agency warns parents of children living in older homes to be aware of high-use areas such as window sills, doors, door frames, stairs, railings, banisters and porches. Lead can disrupt children’s brain development, fine motor skills and emotions. Low levels can cause behavioral problems, loss of IQ and attention deficit disorders. Mental illness symptoms attributed to lead exposure varied significantly by age groups. Those born in 1940 and 2015 had among the lowest levels. Those born between 1966 and 1986 generally had higher mental illness levels linked to lead exposure with the rates peaking for those born between 1966 and 1970, the study said. Those rates coincided with the peak use of lead in gas from the mid-1960s through the mid-1970s.
Lead was added to gas to improve engine performance and eliminate engine knocking. As cars burned the fuel, emissions spread into the atmosphere and accumulated in dust, soil and the lungs of millions of people. Use of leaded gas tapered in the mid-1970s because the fuel┬Ādamaged catalytic converters that became more common to meet stricter emission standards required by the 1970 Clean Air Act. The study said the peak lead use coincided with increased demand for psychiatric care and higher rates of juvenile delinquency. The study cited earlier research detailing the “lead-crime hypothesis” that suggested lead abatement might have contributed to lower murder rates from the 1990s through the 2010s. Now, children are routinely screened for high lead levels in their blood and treated if their levels are concerning. “In the 60s, 70s and 80s, folks were walking around with an average blood lead value that today would trigger clinical follow-up,” Reuben said.”
LEAD-CRIME HYPOTHESIS
https://sciencedirect.com/science/article/pii/S0166046222000667
https://bbc.com/news/magazine-27067615
Did removing lead from petrol spark a decline in crime?
by Dominic Casciani┬Ā /┬Ā 21 April 2014
“Many Western nations have experienced significant declines in crime in recent decades, but could the removal of lead from petrol explain that? Working away in his laboratory in 1921, Thomas Midgley wanted to fuel a brighter tomorrow. He created tetraethyl lead – a compound that would make car engines more efficient than ever. But did the lead that we added to our petrol do something so much worse? Was it the cause of a decades-long crime wave that is only now abating as the poisonous element is removed from our environment? For most of the 20th Century crime rose and rose and rose. Every time a new home secretary took office in the UK – or their equivalents in justice and interior ministries elsewhere – officials would show them graphs and mumble apologetically that there was nothing they could do to stop crime rising. Then, about 20 years ago, the trend reversed – and all the broad measures of key crimes have been falling ever since.

“Thomas Midgley, creator of tetraethyl lead”
Offending has fallen in nations whose governments have implemented completely different policies to their neighbours. If your nation locks up more criminals than the average, crime has fallen. If it locks up fewer… crime has fallen. Nobody seems to know for sure why. But there are some people that believe the removal of lead from petrol was a key factor. Lead can be absorbed into bones, teeth and blood. It causes kidney damage, inhibits body growth, causes abdominal pain, anaemia and can damage the nervous system. More than a century ago, a royal commission recommended to British ministers that women shouldn’t work in lead-related industry because of damage to their reproductive organs. By the 1970s, studies showed that children could even be poisoned by chewing fingernails harbouring tiny flecks of old leaded paint from their homes and schools. Studies have shown that exposure to lead during pregnancy reduces the head circumference of infants. In children and adults, it causes headaches, inhibits IQ and can lead to aggressive or dysfunctional behaviour. If you want to understand the causes of crime – and be tough on them – you need to start with lead, says Dr Bernard Gesch, a physiologist at Oxford University who has studied the effect of diet and other environmental factors on criminals.

“Lead is a very potent neurotoxin,” says Gesch. “It has a range of effects on the brain that have been demonstrated through hundreds of different biological studies. Lead alters the formation of the brain. It reduces the grey matter in areas responsible for things such as impulse control and executive functioning – meaning thinking and planning.” In other words – lead poisoning leads to bad decisions. The lead theorists say the poison has a time-lag effect which could not be understood until recently. In the early 1990s, economist and housing consultant Rick Nevin was pondering whether it would be worth the US spending vast sums cleaning leaded paint out of old housing in American cities. By then, everyone knew that lead did bad stuff to the brain and took years to leave the body. And that got Nevin thinking. Would the accumulation of lead over time play a role in behaviour that would ultimately become criminal? Nevin calculated the rise and fall of the presence of lead from petrol and he compared that curve to the modern history of violent crime. What he came up with was rather startling. When the amount of lead in the environment increased, Nevin showed a corresponding rise in violent crime two decades later.

And when the amount of lead in the environment fell, violent crime also tracked down – again about 20 years later. Was this a one-off, freak statistical result? Where was the proof that lead had actually caused crime? Fourteen years ago, Prof Jessica Wolpaw-Reyes, an economist at Amherst College Massachusetts, was pregnant and doing what many expectant mothers do – learning about the risks to her unborn child’s health. She started to read up on lead in the environment and, like Nevin before her, began pondering its link to crime. “Everyone was trying to understand why crime was going down,” she recalls. “So I wanted to test if there was a causal link between lead and violent crime and the way I did that was to look at the removal of leaded petrol from US states in the 1970s, to see if that could be linked to patterns of crime reduction in the 1990s.” Wolpaw-Reyes gathered lead data from each state, including figures for gasoline sales. She plotted the crime rates in each area and then used common statistical techniques to exclude other factors that could cause crime.
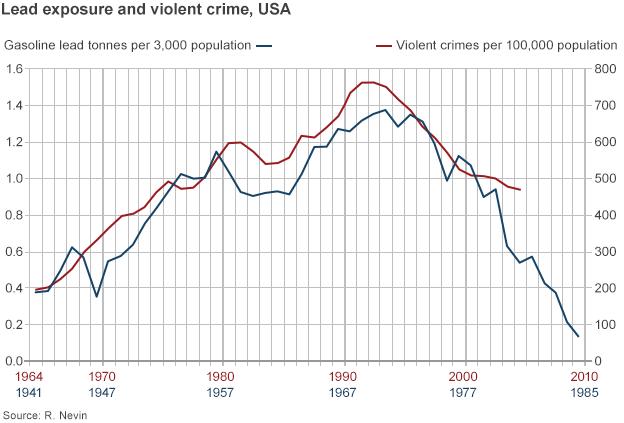
Her results backed the lead-crime hypothesis. “There is a substantial causal relationship,” she says. “I can see it in the state-to-state variations. States that experienced particularly early or particularly sharp declines in lead experienced particularly early or particularly sharp declines in violent crime 20 years later.” She says her research also established different levels of crime in states with high and low lead rates. Nevin’s original research pointed to lead poisoning in childhood increasing the likelihood of offending by the time someone had reached their teens or early twenties. Wolpaw-Reyes’ data appeared to show that anti-pollution legislation in the US then reversed that trend on a state-by-state basis. “Lead changes who we are,” she says. “If you wanted to say, Jessica, I don’t believe that story, then my answer is that you need to come up with another story that would explain why we have found this particular pattern to lead in the 1970s and 80s and then crime in the 1990s and 2000s.
“Moreover you need to be able to show why this relationship is now coming up in other work on bullying, child behaviour problems, teenage delinquency, suicide and substance abuse. You need to tell a story about why those would be linked by chance.” Since then, the data for the lead theorists has become more and more detailed. Nevin and his supporters predicted that crime would fall in other nations 20 years after the banning of leaded petrol – and their theory appears to have played out in Europe. Leaded petrol was removed from British engines later than in North America – and the crime rate in the UK began to fall later than in the US and Canada. Lead theorists say that data they’ve collated and calculated from each nation shows the same 20-year trend – the sooner lead is removed from the environment, the sooner crime will begin to fall.
Dr Bernard Gesch says the data now suggests that lead could account for as much as 90% of the changing crime rate during the 20th Century across all of the world. “A lot of people would say that correlation isn’t cause,” he says. “But it seems that the more the exposure, the more extreme the behaviour. I’m certainly not saying that lead is the only explanation why crime is falling – but it is certainly the most persuasive. Unless someone is telling us that the brain is not involved in decision-making then lead has to be relevant to crime.” So why isn’t this theory universally accepted? Well, it remains a theory because nobody could ever deliberately poison thousands of children to see whether they became criminals later in life. Lead theorists say that doesn’t matter because the big problem is mainstream criminologists and policymakers who can’t think outside the box. But Roger Matthews, professor of criminology at the University of Kent, rejects that. He says biological criminologists completely miss the point. “I don’t see the link,” he says. “If this causes some sort of effect, why should those effects be criminal?
The things that push people into crime are very different kinds of phenomena, not in the nature of their brain tissue. The problem about the theory is that a lot of these [researchers] are not remotely interested or cued into the kinds of things in the mainstream. “There has been a long history of people trying to link biology to crime – that some people have their eyes too close together, or an extra chromosome, or whatever. “This stuff gets disproved and disproved. But it keeps popping up. It’s like a bad penny.” Gesch disagrees and says the debate among biologists is now moving further, to look at how improving nutrition could affect antisocial behaviour. “One of the problems with criminal justice data is that very few of the factors are good at predicting rates of crime. But lead is.” Wolpaw-Reyes says the data is now so good that she is finding that police and policymakers in the US are taking it more seriously. “I don’t think lead is the whole story but given the broad worldwide exposure and the evidence that we have of how adversely lead can affect behaviour, it makes sense that lead is an important part of the story. All of us born in the 60s and 70s were exposed to high levels of lead – but we are not all violent criminals. “But if you had to guess if someone was going to commit a crime I would think that their childhood lead exposure would be helpful to me in making a guess. But it is not going to be a certainty.”
PREVIOUSLY
DEATH by LOONY GAS
https://spectrevision.net/2019/03/14/the-secret-history-of-big-lead/
LEAD and NEURONAL DEATH
https://spectrevision.net/2018/02/22/lead-and-neuronal-death/
ALL LEAD PIPES GO to ROME
https://spectrevision.net/2021/07/07/lead-poisoning-the-roman-empire/
