RAIN RAIN GO AWAY
https://en.wikipedia.org/wiki/Sewer_socialism
“…The policies of Zohran Mamdani have also been compared to sewer socialism. In a speech at the Knockdown Center marking his first 100 days as Mayor of New York City, Mamdani described his administration’s focus on municipal services as “pothole politics“. Mamdani called this approach “our 2026 answer to sewer socialism” and highlighted efforts such as filling 100,000 potholes, increasing garbage containerization, and modernizing catch basins to show how government can deliver public goods.”
COME AGAIN SOME OTHER DAY
https://vitalcitynyc.org/floodproofing-new-york-city/
https://vitalcitynyc.org/nyc-floodproofing-cloudburst-rain-policy/
Flooding in New York is a Policy Choice
by Amy Chester┬Ā&┬ĀTyler Taba / July 22 2026
“The City can and must guard against the consequences of extreme rainfall. After a week of torrid temperatures and wildfire smoke, New Yorkers woke up to another climate disaster ŌĆö┬Āflash flooding on July 18. If it felt familiar, thatŌĆÖs because it was. Just two months ago, on May 20, a flash flood paralyzed much of the city. During that storm, New York CityŌĆÖs┬ĀFloodNet sensor network ŌĆö a series of roughly 431 flood sensors managed by NYU and CUNY ŌĆö detected flooding across 98 sensors. One sensor in┬ĀHollis, Queens recorded 46 inches of street-level flood water┬Āin under two hours. This time around, a sensor in┬ĀFresh Meadows, Queens recorded 29.3 inches in about an hour. ItŌĆÖs not actually the total amount of rainfall and street-level flood depths that are the most alarming; rather, itŌĆÖs the sudden deluge in a very short period of time. Meteorologists have confirmed that in the worst-case scenarios, the brunt of the rainfall can come down in just 20 to 40 minutes. That is truly the epitome of what we call a ŌĆ£cloudburstŌĆØ event.
Believe it or not, New York is better equipped to handle flash flooding than it was 10 or 20 years ago. After Hurricane Ida in 2011, the CityŌĆÖs Department of Environmental Protection (DEP) made significant investments in a Cloudburst Management program that aims to alleviate the impacts from these sudden, heavy downpours. The most famous example is the award-winning sunken basketball court, modeled after similar projects in the Netherlands, at NYCHAŌĆÖs South Jamaica Houses. Building on that work, a series of Cloudburst projects were announced in Corona, Kissena Park, Parkchester and East New York, followed by a recent $95 million investment from Mayor Mamdani in Homecrest, Brooklyn earlier this year.
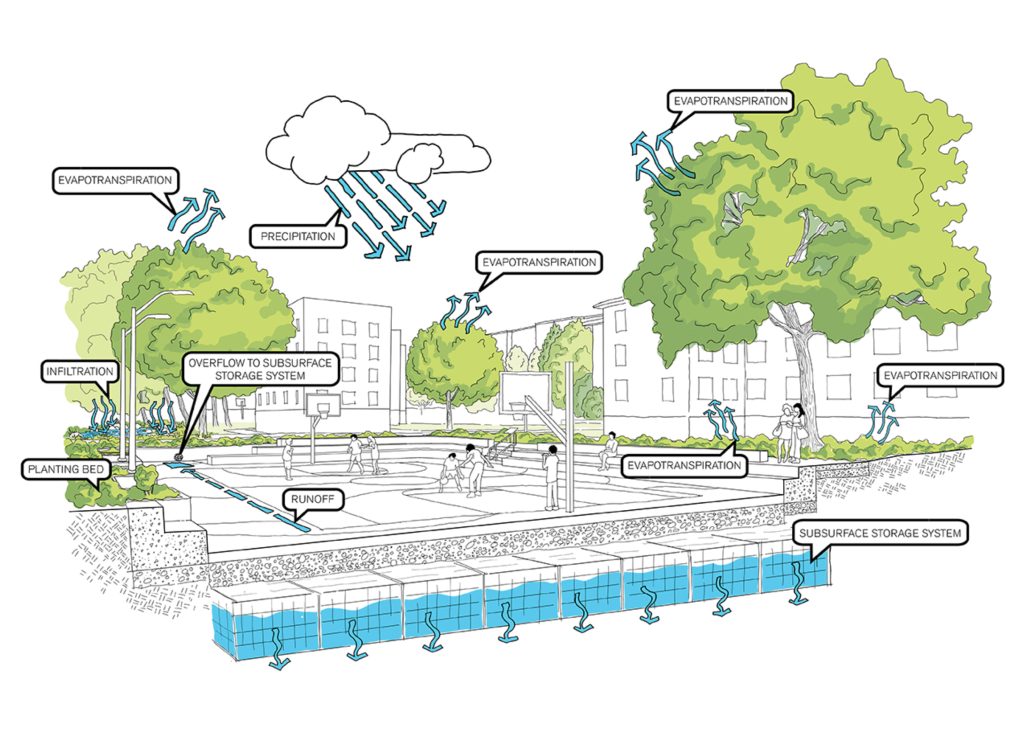
Communities across New York City are also taking it upon themselves to prepare their communities and neighbors for the next big storm. In Gowanus, artist Tiffany Baker is transforming flood-impact stories from Gowanus residents into an outdoor mural campaign accompanied by practical flood prevention education. In Hollis, the Southeast Queens Residents Environmental Justice Coalition is documenting the story┬Āof flooding in parts of the neighborhood that have no sewers. In Bed-Stuy, the First Nigerian SDA Church trains┬Ālocal youth┬Āto perform peer education across the community. There is even a flood preparedness┬Āplay┬Āthat has been performed in chai shops and parks throughout the city, and the development of an AI chatbot for renters. It all amounts to real progress ŌĆö but itŌĆÖs not nearly enough.
In fact, allowing our public transit system, roads, homes, and businesses to flood like they do is a policy choice. The CityŌĆÖs sewer system is designed to handle 1.75 inches of rainfall per hour, so when we see 2 inches in a mere 20 minutes, floodwaters will naturally travel to the nearest low-lying area, which is often street ends and basements. At DEPŌĆÖs current pace, which is roughly $1 billion per year, it would take approximately┬Ā30 years┬Āto implement capital improvements in the 80+ stormwater priority areas they have identified. A full build-out to modernize our system would cost upwards of $250 billion. In the meantime, New Yorkers end up footing the bill for lost wages due to disrupted commutes, property damages and long-term health conditions from toxic water and mold.

Mayor Mamdani can reduce these impacts by emboldening every city agency to capture, absorb and store water where they can. In his announcement of the CityŌĆÖs┬Ānewest Cloudburst project, the Mayor made it clear that ŌĆ£as climate change accelerates, investments like this are not optional. They are how we deliver a city that is resilient, equitable and prepared.ŌĆØ There are promising signs of MamdaniŌĆÖs mandate across some city agencies. The Department of Transportation has been unleashed to upgrade and improve the network of bike and bus lanes. The Department of Consumer and Worker Protection has adopted an aggressive enforcement posture to protect consumers and gig workers.
Alongside strong city agency leadership, the mayor should embrace the power of community leadership and visioning. In response to Hurricane Ida, Rebuild by Design and One Architecture & Urbanism convened New Yorkers representing 16 government agencies, 20+ non-profits, 12 neighborhood organizations and 10+ academic, private or philanthropic institutions. That convening, known as Rainproof NYC, was a six-month process aimed at developing recommendations to address the increasing flooding from heavy rainfall in New York City. Mayor Eric Adams largely shelved those recommendations. Now, as we are into his successorŌĆÖs first cloudburst season, which typically runs from July through September, it might be time for some literal sewer socialism.
HereŌĆÖs what that would look like. First, there needs to be a clear leader on ŌĆ£rainproofingŌĆØ New York City ŌĆö┬Āsomeone [like Kathryn Garcia?] with the authority to convene an interagency task force that tracks metrics and holds each city agency accountable for their role. The burden of climate adaptation cannot fall on DEP and New York City Emergency Management (NYCEM) alone. Instead, the City must orchestrate an all-agency approach that should begin with an inventory of communications assets across every agency to determine how the City can educate and prepare New Yorkers, especially on dry days.
Then comes infrastructure, starting with City-owned property. The late Chinese landscape architect, Kongjian Yu, inspired an entire generation with the┬Ā“sponge city” concept. By replacing impervious surfaces with rain gardens, green roofs and urban wetlands, and by using landscapes to slow down and store stormwater, sponge cities challenge us to reimagine our relationship with water. To embrace that challenge, the City should utilize every piece of property it owns to delay and store water. ThatŌĆÖs 362 million square feet of land across nearly 5,000 parcels. Streets, public plazas and buildings should all be part of a network of green infrastructure that works to safely drain stormwater before it becomes floodwater.
Perhaps the best places to start are where other benefits can be leveraged. For example,┬ĀNew York City parks┬Āface the compounding risks of high flood vulnerability, high heat vulnerability and high social vulnerability. Across the Hudson River,┬Āthe City of Hoboken┬Āhas successfully lowered flooding by 88% by investing in resilient parks. This can be replicated in every neighborhood. Creating sophisticated infrastructure to reroute water from surrounding streets to underneath our parks would be a win-win situation, reducing the chances of flooding while creating well-paying, local green jobs. Mayor MamdaniŌĆÖs latest┬Ā$4.5 million green infrastructure workforce pilot┬Āin East New York and South Ozone Park is a great start to the green jobs pipeline.
At the same time, we have to harden the subways, which flood chronically, against extreme rainfall. The MTA is making an investment in flood mitigation, but they need massive resources to shore up the whole system. You may have seen examples of this such as elevating street-level stairs ŌĆö having the rider step up two steps, to go down ŌĆö and covering ventilation grates on the sidewalks so floodwater doesn’t enter from above. It works, but given the scope of the challenge, weŌĆÖve only seen changes around the margins so far. Private property, including homes and businesses, also has a critical role to play in flood prevention.
To encourage more on-site capture, New York City should create a new incentive program, similar to Philadelphia’s┬ĀRain Check┬Āor Washington D.C.’s┬ĀRiverSmart program, which can be tailored for private properties in under-resourced and high flood risk neighborhoods. Lastly, on the neighborhood scale, the research is clear that┬Āsocial infrastructure┬ĀŌĆō the strength of the connections neighbors have to one another ŌĆō can be a life-or-death determinant in severe weather events. By collaborating with community leaders to educate local residents, the City can build capacity block by block to help New Yorkers survive these increasing disruptions. It is possible to imagine a greener, more resilient, and safer city ŌĆö even when it rains. Now it’s up to the Mamdani administration to make it happen.”
PREVIOUSLY
SPONGE CITIES
https://spectrevision.net/2024/06/26/sponge-cities/
FLASH FLOOD SEASON
https://spectrevision.net/2023/09/13/flash-flood-season/
DAYLIGHTING BURIED WATERWAYS
https://spectrevision.net/2022/01/17/daylighting/
MAKING ROOM for RIVERS
https://spectrevision.net/2020/02/02/underwater-urban-planning/
HURRICANE MODIFICATION
https://spectrevision.net/2019/09/05/hurricane-modification/
STORMWATER MANAGEMENT
https://spectrevision.net/2017/09/07/paved-paradise/
