GLUTAMATE CASCADES
https://en.wikipedia.org/wiki/Excitotoxicity
“Excitotoxicity is the pathological process by which nerve cells are damaged or killed by excessive stimulation by neurotransmitters such as glutamate and similar substances. This occurs when receptors for the excitatory neurotransmitter glutamate (glutamate receptors) are overactivated by glutamatergic storm.”
https://metabolichealing.com/glutamate-excitotoxicity-the-brain-the-nervous-system-nutritional-solutions/
“One of the key mechanisms of glutamate-induced neuronal cell injury is in the release of glutamate by microglial cells (13, 14). Microglial cells are essentially the resident immune cells of the brain and nervous system. Once the free pool of glutamate increases, glutamate receptors, such as NMDA are over-stimulated. This then causes a massive influx of calcium ions into cells, resulting in neuronal cell damage, and cell death.”
http://www.truthinlabeling.org/Glut.html
“Reading these studies, the reader will find discussion of damaged or overly sensitive glutamate receptors, and malfunctioning glutamate transport. The reader will also find discussion of “glutamate pools” where excess glutamate is stored – and then sometimes released to cause brain damage. Rarely mentioned, however, is the role that ingestion of excess amounts of processed free glutamate (MSG) might play in producing these diseases; and if mentioned at all, it is by researchers outside of the United States.”
NEURONAL DEATH
http://www.ajnr.org/content/22/10/1813
“Neuroradiologists may encounter, on a daily basis, a challenging diversity of neurologic disorders, including stroke, trauma, epilepsy, and even neurodegenerative conditions, such as Huntington disease, AIDS dementia complex, and amyotrophic lateral sclerosis (1), but this spectrum of disease is not usually thought of as sharing the same mechanism of neuronal injury and death. These and a growing list of other neurologic disorders are now understood to share a final common destructive metabolic pathway called excitotoxicity (2, 3), which has been the focus of intense investigative efforts in the neurosciences over the past several decades (3–31).
Excitotoxicity refers to an excessive activation of neuronal amino acid receptors. The specific type of excitotoxicity triggered by the amino acid glutamate is the key mechanism implicated in the mediation of neuronal death in many disorders. The discovery of excitotoxic injury is a major clue in the search for answers to such fundamental questions as why neurons die in disease states and what is the precise or critical mechanism of neuronal death.
This overview introduces and reviews some of the major concepts of glutamate excitotoxicity. Familiarity with this intriguing pathologic process will enhance the understanding of the neuroimaging changes of many neuropathologic processes, facilitate a conceptual model for some of the newer treatment strategies for some disorders (ie, stroke and trauma), and perhaps cultivate new directions for neuroimaging. The organization of this article follows the excitotoxic process from the formation of glutamate to neuronal death.
Glutamate excitotoxicity is a broad and rapidly evolving field of study with many important nuances that have necessarily been oversimplified or, unfortunately, omitted from this review for the sake of reasonable brevity. The reader is referred to the exponentially expanding literature and some of the sources listed in the reference section for additional information.
https://www.youtube.com/watch?v=uS0ioiB0_oY
Glutamate
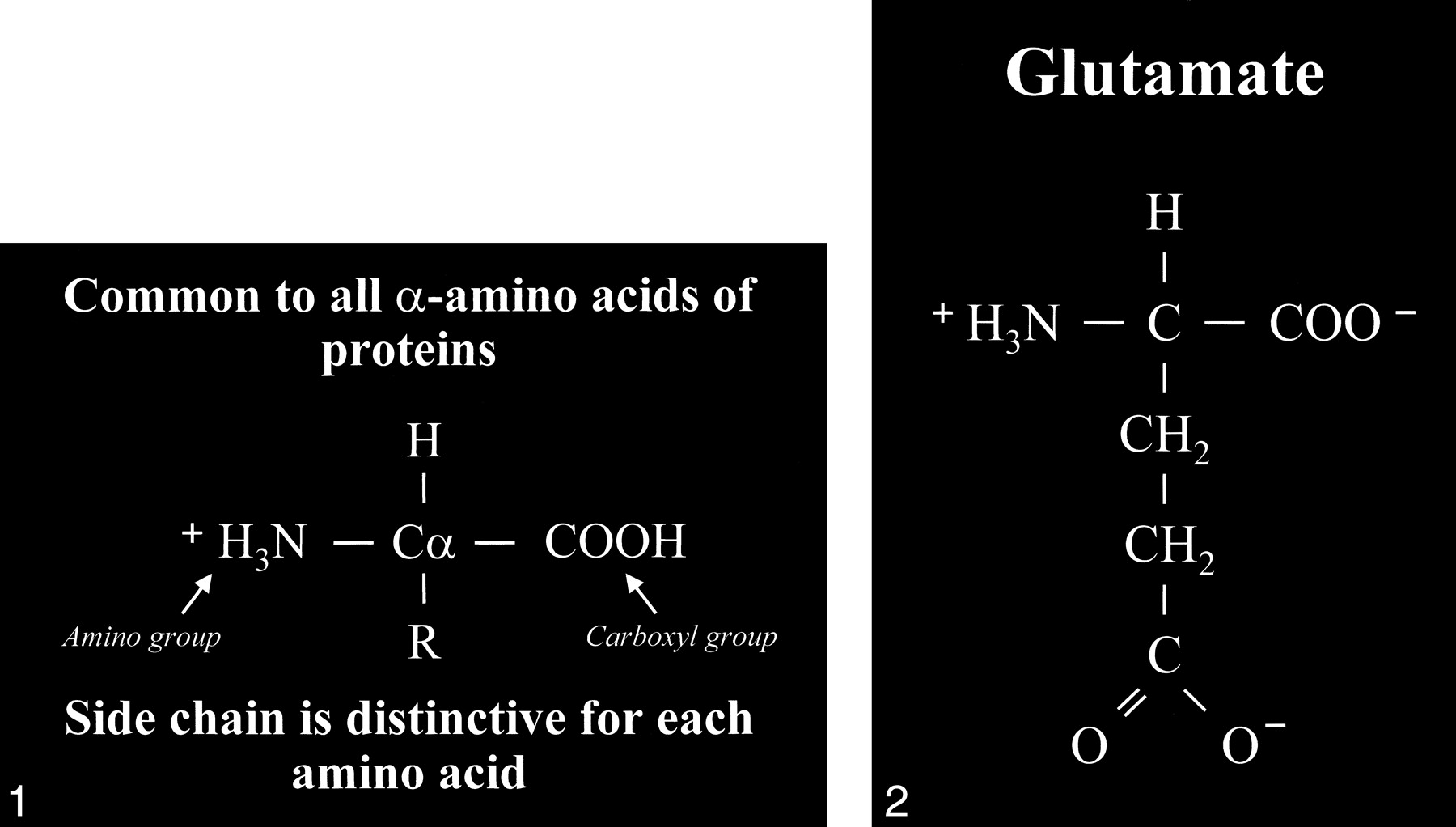
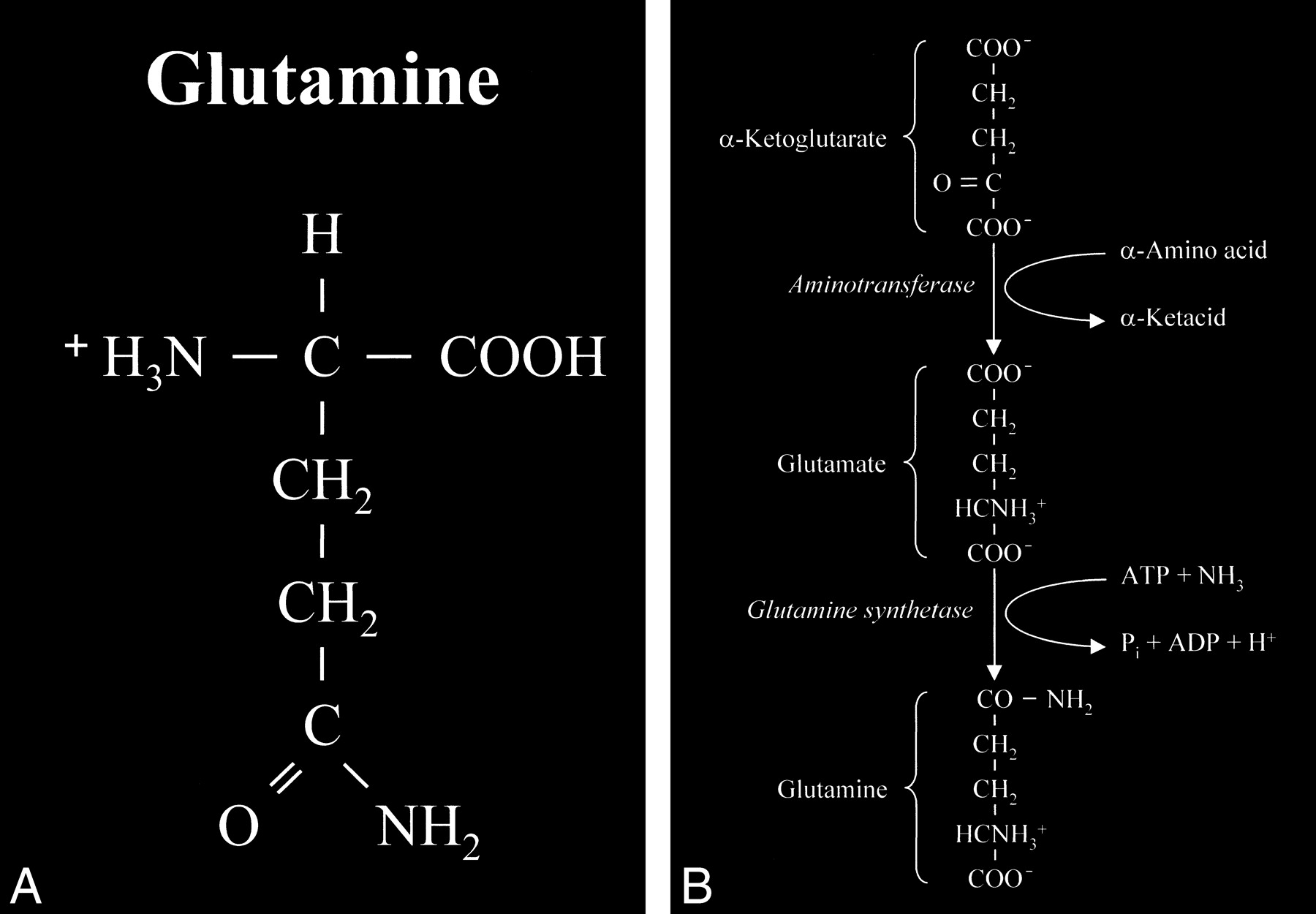
Glutamate is an amino acid and one of a group of amino acid neurotransmitters in the brain, although it is the principal excitatory neurotransmitter. More basically, amino acids (Fig 1) consist of a central carbon atom (α carbon) bonded to a carboxyl group (COOH) and an amino group (NH3). A distinctive side chain (R group), which characterizes each amino acid, links to the α carbon. Glutamate (Fig 2) consists of the side chain CH2CH2COO– (COOH ending [γ carboxyl group] for glutamic acid) attached to the α carbon, while the closely related glutamine (Fig 3A and B) is created from glutamate with ammonia added at the γ carboxyl group by glutamine synthetase, forming the CH2CH2CONH2 side chain R group. Cerebral glutamate is derived solely from endogenous sources; mainly from α ketoglutarate, which is a product of the Krebs cycle (citric acid cycle, TCA [tricarboxylic acid] cycle).

Download figure / Download powerpoint
fig 1. Amino acid. Typical amino acid consists of a central alpha carbon (Cα) that is bonded to a carboxyl group (COOH) on one side and an amino group (H3N) on the other side. Each amino acid is characterized by a distinctive molecular group (R) or side chain attached to the α carbon.
fig 2. Glutamate. The side chain, or R group, of glutamic acid is CH2CH2COOH. The carboxyl group of the side chain is designated the γ carboxyl group, which becomes fully ionized at neutral pH and is therefore frequently written with a negative charge (COO−). The term glutamate (instead of glutamic acid) is used to indicate this negative charge or ionized state at physiological pH.

Download figure / Download powerpoint
fig 3. A, Glutamine. This closely related amino acid is formed from glutamate with the addition of an amino group at the γ carboxyl of the side chain. B, This formation of an amide linkage at the γ carboxyl group requires the enzyme glutamine synthetase and the process is adenosine triphosphate (ATP)-dependent. This reaction is also a major mechanism for the detoxification of cerebral ammonia.
The neuronal glutamate considered here acts as a neurotransmitter, which is the method of communication between neurons. This interaction between neurons may be either excitatory or inhibitory. The major excitatory amino acid neurotransmitters are glutamate and aspartate, while GABA (γ-aminobutyric acid), glycine (aminoacetic acid), and taurine are inhibitory.
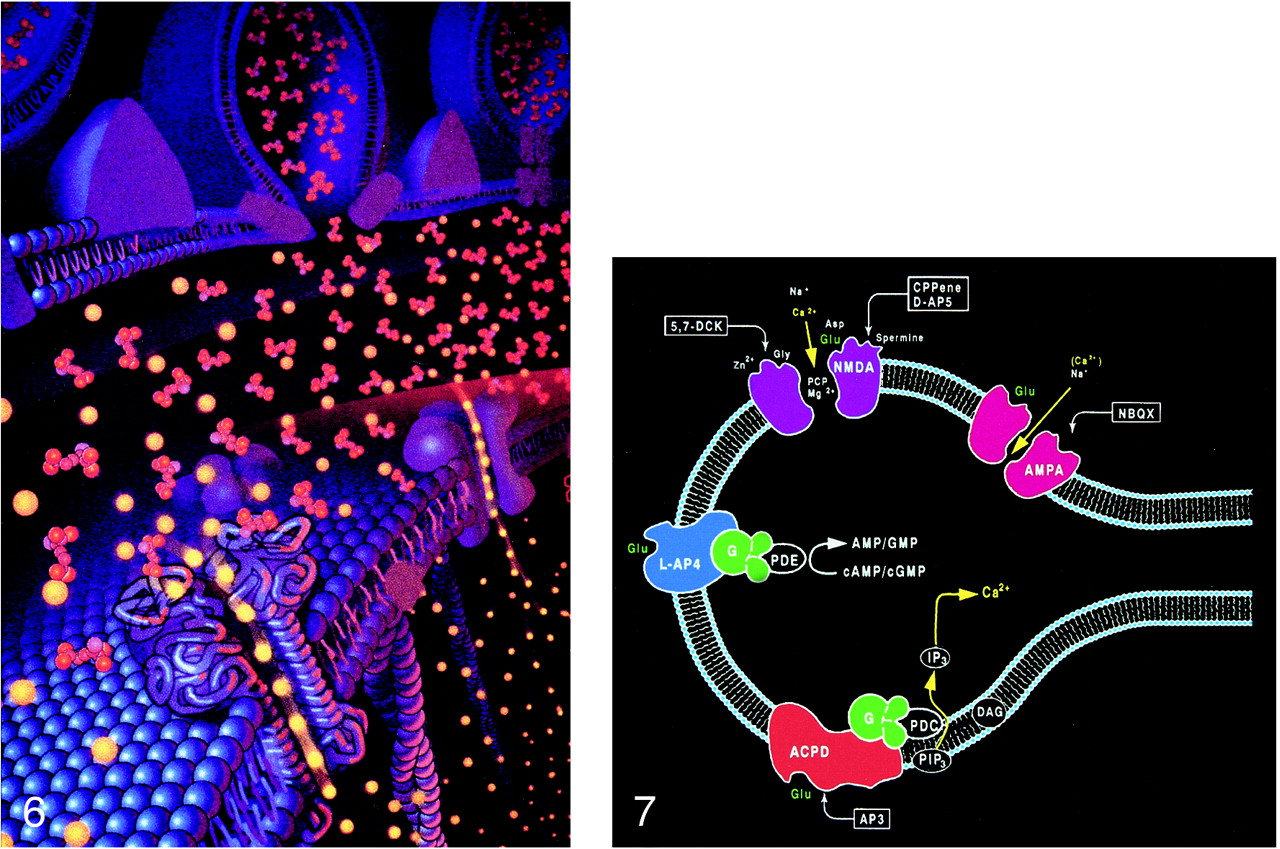
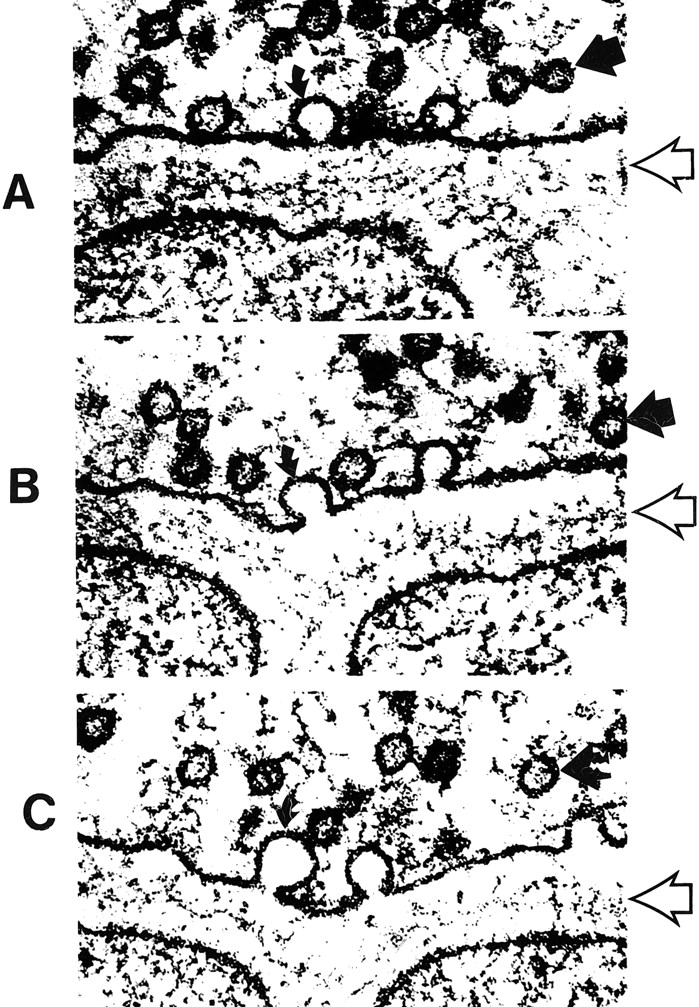
The processing and transport of glutamate (Fig 4) within the neuron are highly organized and coordinated interactions among multiple cytoplasmic organelles resembling the frenetically detailed but choreographed mosaic activities of an ant farm (32). Glutamate, like other neurosecretory substances, is initially synthesized by the endoplasmic reticulum and then transported to the Golgi apparatus for additional processing. Emerging from the opposite surface of the Golgi apparatus and wrapped inside a vesicular (bilipid) membrane, glutamate is then transported down the axon via a complex system of microtubules. Antegrade motion down the axon on the microtubules is mediated by molecules called motor kinesin, whereas cytoplasmic dynein generates retrograde motion. Mitochondria also accompany these transport molecules, providing the required energy. Upon reaching the axonal tip (Fig 5), the vesicle with the enclosed glutamate merges with the presynaptic membrane by the process called exocytosis to release the glutamate into the synaptic space between neurons. The vesicular membrane is then recycled and transported back up the neuronal axon in a retrograde fashion via the microtubular network. The synaptic glutamate is finally freed to interact with specific receptor sites on the postsynaptic membrane of the adjacent neuron to initiate an important cascade of molecular events within that neuron (Fig 6).

Download figure / Download powerpoint
fig 4. Neuronal glutamate processing and transport. Glutamate is processed by the endoplasmic reticulum and Golgi apparatus in preparation for fast axonal transport, which also requires other transport proteins and mitochondria. When glutamate emerges from the “trans” face of the Golgi apparatus, it is encapsulated inside a neurosecretory vesicle, which consists of a bilipid membrane. These vesicles are transported down the axon along microtubule tracks to be deposited at the tip of the axon near the presynaptic membrane. Waves of axonal membrane depolarization would trigger the release of the glutamate into the synaptic space by exocytosis, which is exhibited by the merging of the neurosecretory vesicles with the postsynaptic membrane to free the packaged glutamate. The empty vesicle would then be recycled back to the neuronal body by retrograde transport along the microtubular tracks (adapted from [32])

Download figure / Download powerpoint
fig 5. A–C,Electron micrographs show neurosecretory vesicles releasing neurotransmitter molecules by exocytosis on the presynaptic membrane. Numerous small neurotransmitter substances can be seen in the synaptic space (open arrows). These neurotransmitters will then settle on and activate receptors on the postsynaptic membrane. Dark circles by straight solid arrows represent vesicles filled with neurotransmitters. Light partial circles by curved solid arrows represent the vesicle merging with the presynaptic membrane and releasing neurotransmitter into the synaptic space (adapted from [32])
Download figure / Download powerpoint
fig 6. Pictorial display of the neurotransmitter glutamate (orange) released into the synaptic space and docking with the glutamate receptor site on the postsynaptic membrane. The activation of the glutamate receptor then opens the ion channel coupled to the receptor, allowing the passage of extracellular calcium (yellow) into the intracellular cytosol, which in turn triggers a series of biochemical events (adapted from Schornak S, BNI Q 11:1995)
fig 7. Ionotropic and metabotropic receptors. The ionotropic receptors NMDA (purple) and AMPA (red) are directly coupled to ion channels. The metabotropic receptors (blue and orange) activate intermediary molecules such as G protein affecting multiple cytoplasmic enzymes to produce molecules, such as IP3, that increase cytosol calcium concentrations. Also depicted are modulatory substances, such as spermine, which facilitate calcium influx, and receptor complex inhibitors, such as zinc, magnesium, and PCP. L-2-amino-4-phosphonopriopionic acid (L-AP4) and aminocyclopentyl dicarboxylic acid (ACPD) receptors are classified as metabotropic, as they are coupled to intermediary G proteins (G) that activate phosphodiesterase (PDE) for L-AP4 receptors and form inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol 4,5-bisphosphate (PIP3) for the ACPD receptors via phospholipase C. 2-amino-3-phosphonopriopionic acid (AP3) and quinoxaline-2,3-dione (NBQX) are antagonists for ACPD and AMPA receptors, respectively.
Postsynaptic Anatomy
The interaction of glutamate with the postsynaptic membrane requires a review of glutamate receptors (Fig 7). The two main types of glutamate receptors are ionotropic and metabotropic. Ionotropic receptors are directly coupled to membrane ion channels. The metabotropic receptors are coupled to intermediary compounds, such as G protein, and modulate intracellular second messengers, such as inositol-1,4,5-trisphosphate (IP3), calcium, and cyclic nucleotides. The directly coupled ionotropic receptor, which is the primary consideration of this review, can be further subdivided into three subtypes: NMDA (N-methyl-D-aspartate), AMPA (a-amino-3-hydroxy-5-methyl-4-isoxazolepropionate), and kainate. These subtypes are named for their selective chemical agonists, which resemble glutamate but do not naturally exist in the brain. This nomenclature may seem awkward, but it offers the convenience and consistency of grouping the various cerebral and cerebellar receptors according to their responses to the chemical tools that are used to evaluate or stimulate them.
Excessive accumulation of intracellular calcium is the key observed process leading to neuronal death or injury, and the NMDA receptors activate channels that allow the influx of extracellular calcium (and sodium). Overstimulation of this type of glutamate receptor would then lead to neuronal calcium overload. Some types of AMPA and kainate receptors can contribute to intracellular calcium overload because their coupled membrane ion channels are at least partially permeable to calcium.
The influx of calcium and sodium from glutamate receptor stimulation results in membrane depolarization, which can also activate voltage-dependent calcium channels. These other calcium channels then allow further calcium influx, aggravating the intracellular calcium overload initiated by overstimulation of the glutamate receptors and opening of the associated ion channels. The four main types of voltage-dependent calcium channels considered here are named for their specific properties: T (transient current), N (found in neurons), L (long duration current, large conductance channels), and P (found in Purkinje cells of the cerebellum). The L channel is not the most prevalent type but it disproportionately contributes to calcium-mediated neuronal injury because of the prolonged calcium influx that occurs with activation of this voltage-dependent conduit.
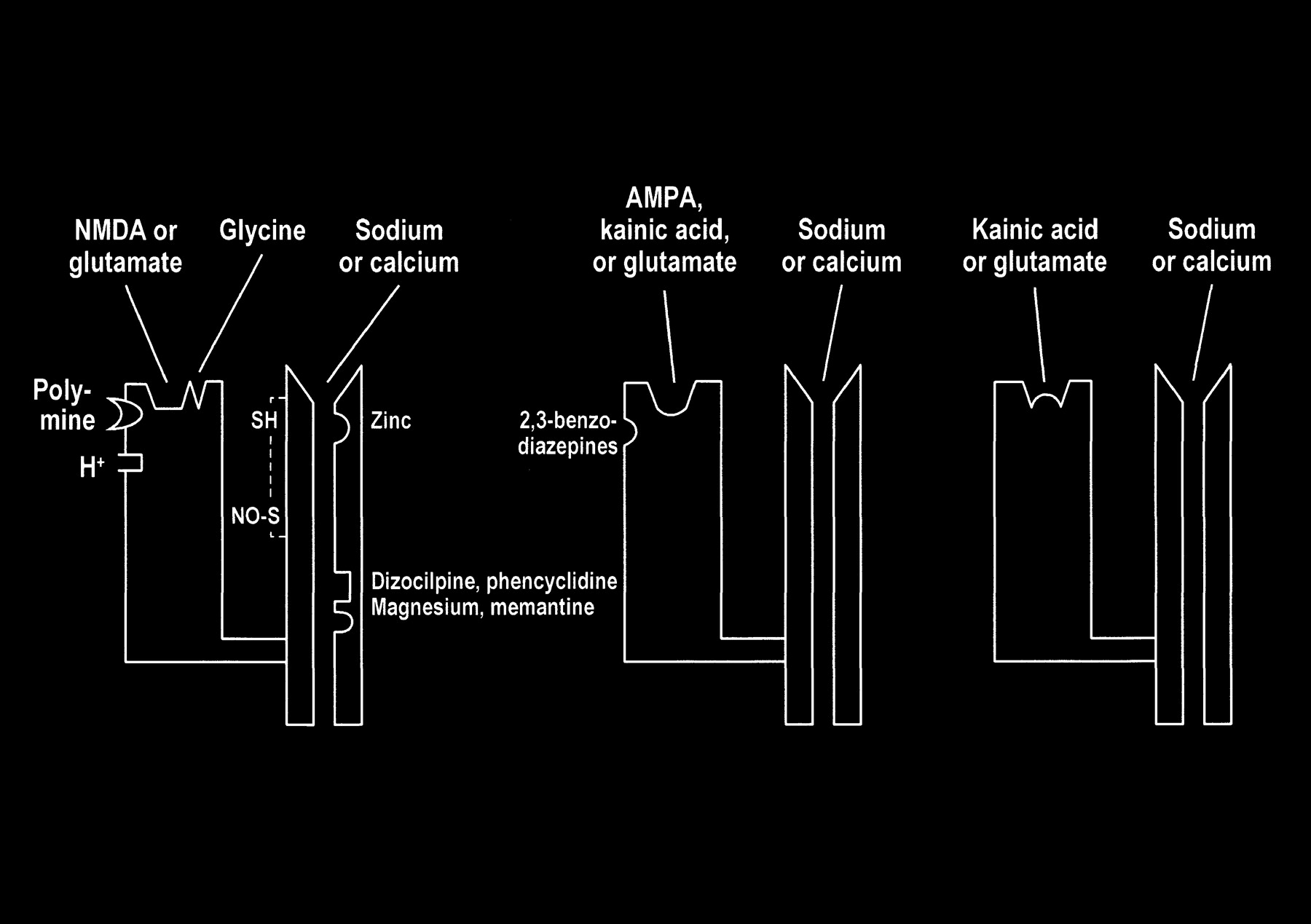
Multiple modulatory sites, however, complicate some of the ionotropic receptors. It may be helpful to think of the receptors as a receptor-channel complex with the receptor closely linked to and controlling the adjacent ion channel. Modulatory sites are separate areas on the receptor and the channel in which other molecules can influence the function of the receptor site or channel. On the NMDA receptors, glycine (once considered a laboratory contaminant) acts as a required coagonist. Hydrogen ions (a reflection of pH) suppress receptor activation. Polyamines, such as spermine, however, can relieve proton block and potentiate NMDA receptor activation in a pH-dependent fashion. The NMDA receptor channels are affected by multiple factors, including magnesium (which blocks the channel), zinc (positive and negative modulator), and multiple drugs, such as dizocilpine and phencyclidine ([PCP, “angel dust”] channel blocker) (Fig 8).

Download figure / Download powerpoint
fig 8. Schematic representation of NMDA, AMPA, and kainate receptors as receptor–channel complexes. Glutamate docks with the receptor, which opens the coupled channel to allow the intracellular influx of extracellular calcium. Other molecules (such as magnesium, zinc, and PCP) can influence receptor function by interacting with several receptor and channel modulatory sites
Calcium
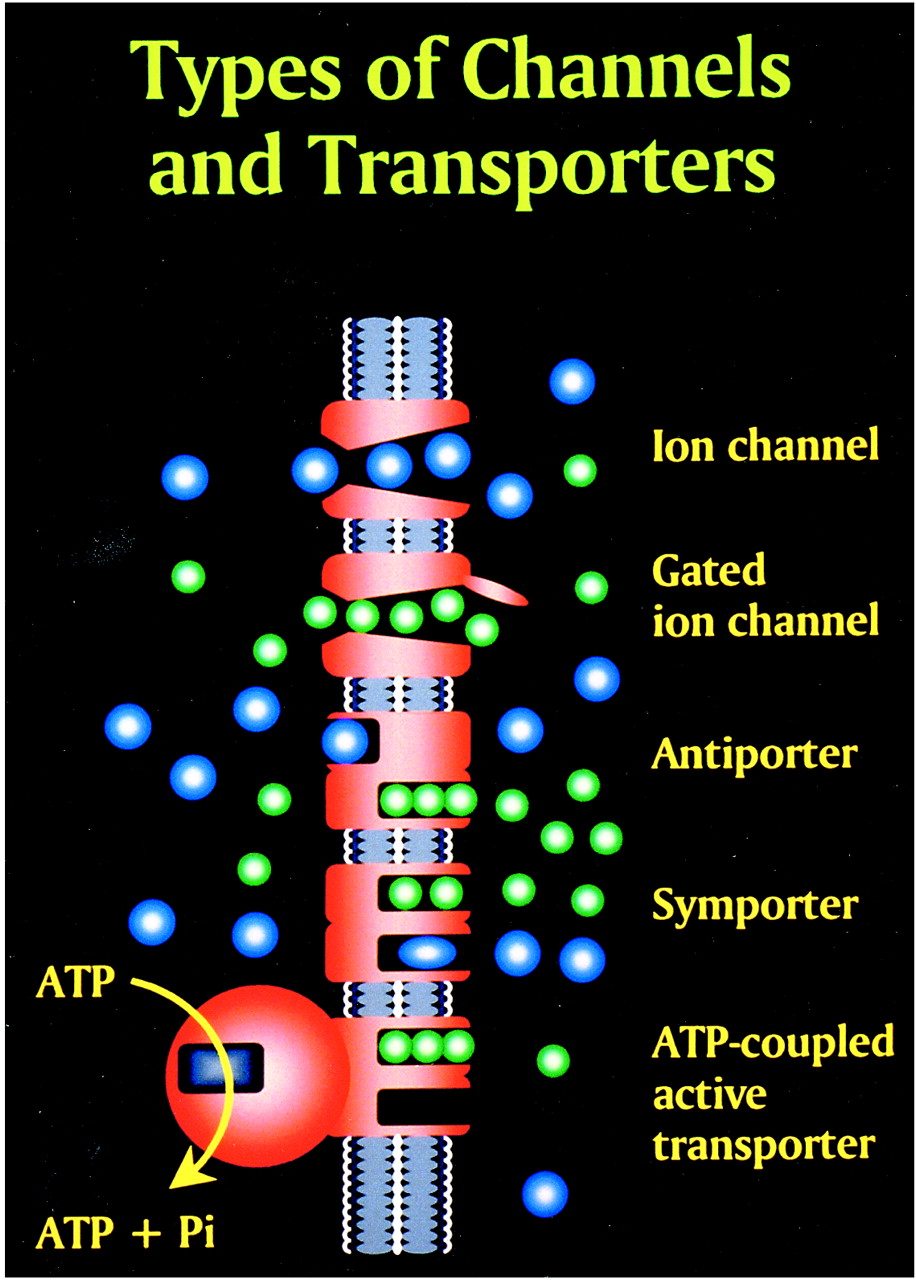
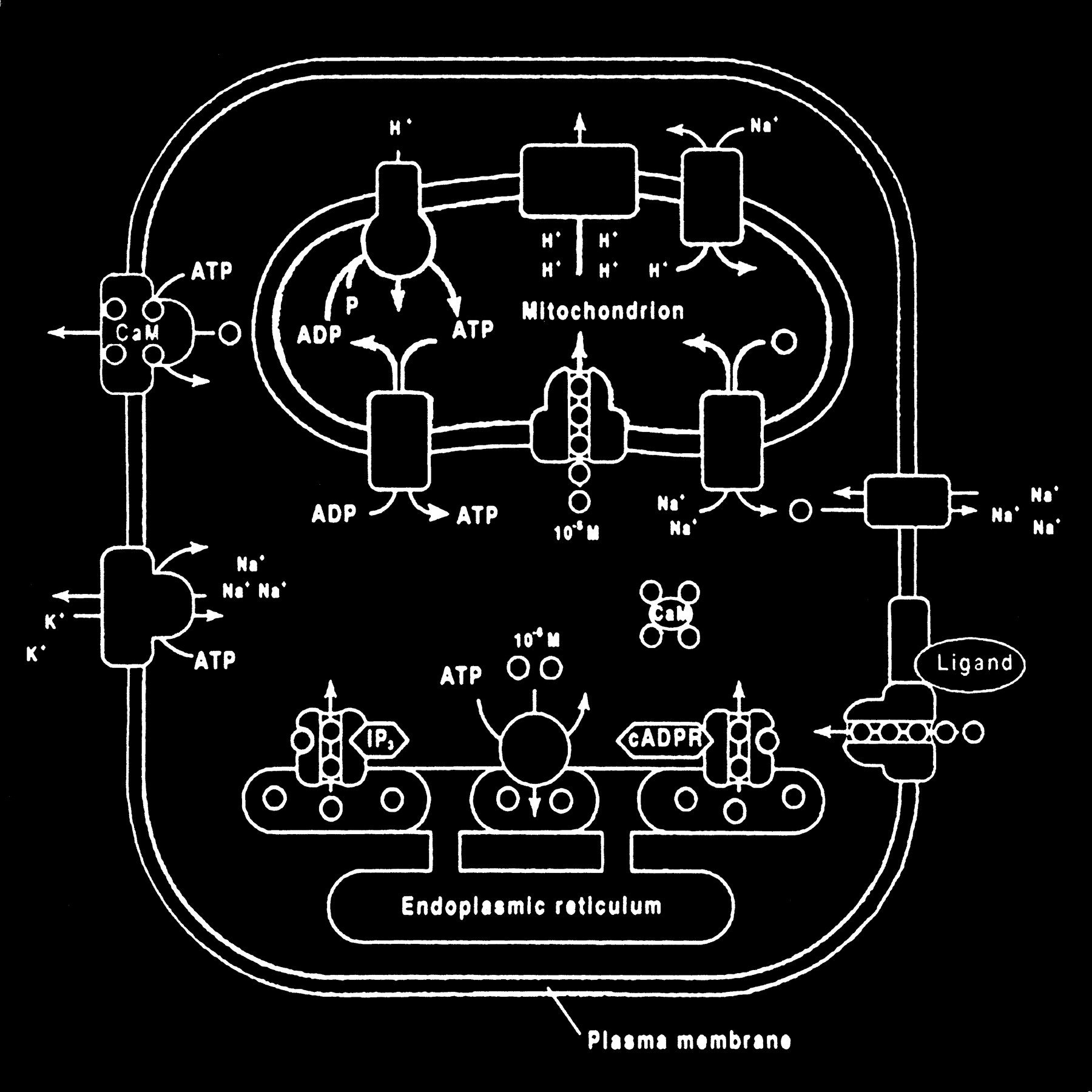
Glutamate excitotoxic calcium overload can be appreciated from the perspective of the normal mechanisms of neuronal calcium homeostasis. Intracellular cytosolic free calcium is maintained at very low concentrations (micromolar) relative to free extracellular calcium. Plasma membrane calcium transporters regulate this cytosolic free calcium concentration. Membrane transporters in general have been classified as antiporters, symporters, and ATP-coupled active transporters (Fig 9). These transporters are membrane protein compounds that are coupled to energy sources and change the distribution of substrate ions or molecules across a membrane. Antiporters and symporters are called secondary transporters because they use the energy from an existing ion gradient to drive the passage of another ion or molecule in the same (symporter) or opposite (antiporter) direction across a membrane as the energizing ion. Movement through the neuronal membrane is achieved by a change in the conformation of the protein-substrate complex. Complexing with two or more substrates is required to initiate conformational transitions in antiporters (opposite direction coupling) and symporters (same direction coupling). The primary transporters (ATP-coupled active transporter) couple a chemical reaction to the protein conformational transitions that supply the metabolic energy required to generate concentration gradients of substrate ions across the membrane. Calcium is controlled by the antiporter and plasma-membrane calcium pump (PMCA). The antiporter, which has a low affinity but high transport capacity for calcium, moves calcium out of the neuron by a sodium–calcium exchange mechanism. The sodium gradient across the membrane drives this exchanger. The PMCA, on the other hand, has a high affinity but low transport capacity for calcium. This active pump transports one Ca2+ for each ATP hydrolyzed. One distinguishing feature of the PMCA is the enhanced activation of the pump by binding Ca2+/calmodulin, which results in a 20- to 30-fold increase in the affinity of the substrate Ca2+ site (33).
Download figure / Download powerpoint
fig 9. Membrane channels and transporters. The ion channels are pathways to allow the diffusion of ions across the cell membrane. These channels can be opened or closed by changes in membrane voltage, associated ligands, and so on. Excessive intracellular calcium is detrimental to neuronal health, and a calcium gradient is maintained across the neuronal membrane mostly by three main types of transporters. The antiporters use an existing ion gradient (usually established by active or ATP-dependent transport) to transport calcium ions (green) in the opposite direction of the energizing ion (ie, sodium). Symporters transport calcium in the same direction of the energizing ion (blue). Both antiporter and symporters are considered secondary transporters because they derive energy from an existing gradient. The ATP-coupled active transporter is considered a primary transporter that uses ATP to affect the transmembrane movement of calcium to establish its gradient (adapted from [32])
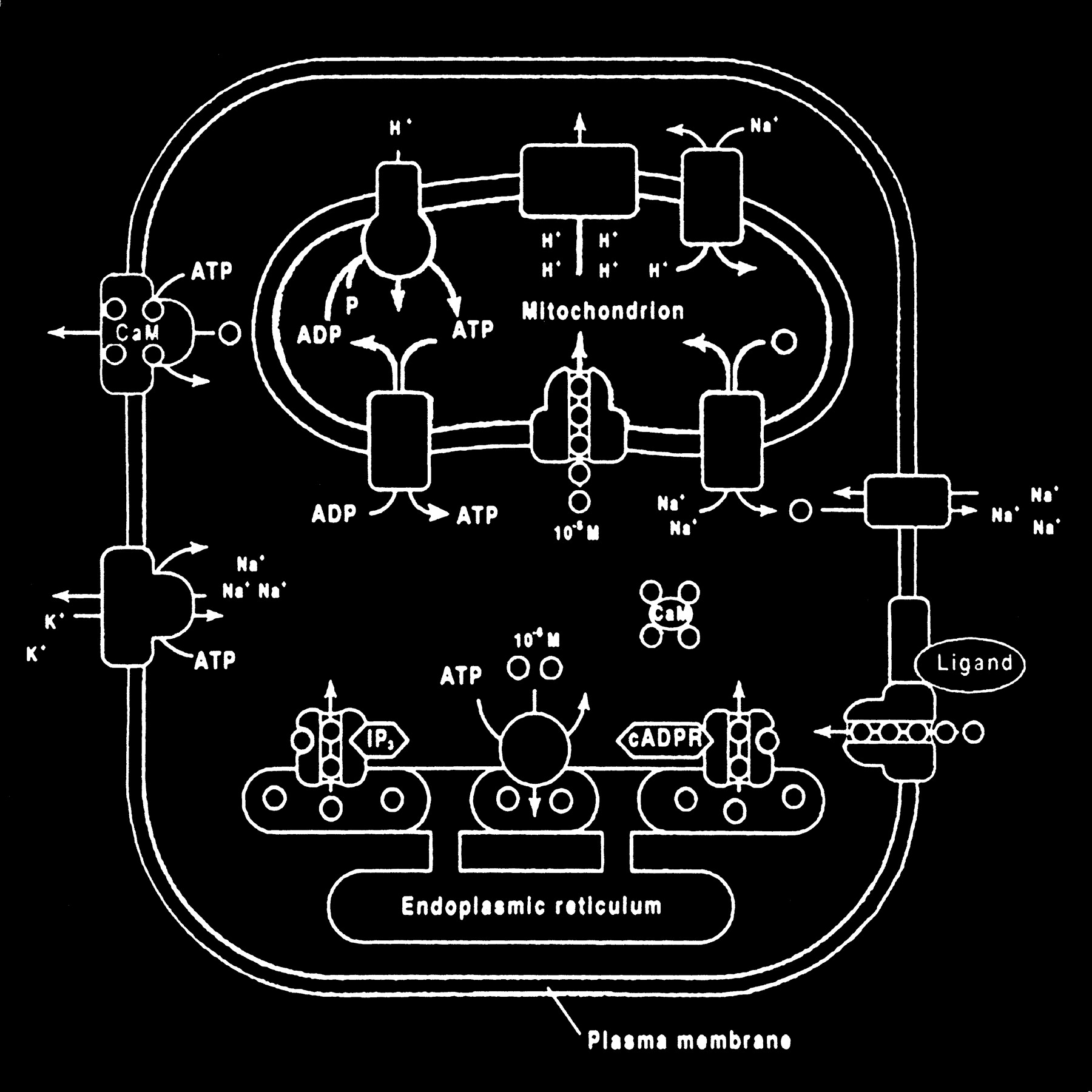
The mitochondrion and endoplasmic reticulum are also significant sources of calcium stores. An antiporter mechanism maintains high calcium concentrations in the mitochondria by moving free calcium from the cytosol to the mitochondria while an antiporter and an ATP-dependent active pump sequester the endoplasmic reticulum calcium (Fig 10).
Download figure / Download powerpoint
fig 10. Calcium homeostasis. Membrane transporter (antiporter and ATP-dependent transporter) maintain a much higher extracellular calcium (small circles) concentration than the cytosol. The endoplasmic reticulum and mitochondria are important sources of intracellular extracytosolic calcium. These internal sources of calcium can be released into the cytosol when provoked by specific agents, such as the second messenger (IP3) actions on the endoplasmic reticulum (adapted from [32])
Glutamate receptor overstimulation increases intracellular calcium by directly opening ion channels and secondarily affecting calcium homeostatic mechanisms. As mentioned, the initial glutamate receptor opening of the sodium/calcium channels not only allows the influx of calcium but also causes membrane depolarization. The depolarization would in turn activate the voltage-dependent calcium channels, which would further increase the intracellular calcium levels. The decreased sodium gradient across the cell membrane caused by the glutamate receptor–coupled channels, however, would also reduce the ability of the sodium gradient–dependent antiporter to remove intracellular calcium. Superimposed disorders that decrease ATP production (ie, hypoxia, neurodegenerative disorders, etc) would adversely affect the activity of the ATP-dependent calcium transporters as well as the energy-dependent sodium potassium pump, which would then also affect the transmembrane sodium gradient and therefore the antiporter function.
Intracellular Toxic Events
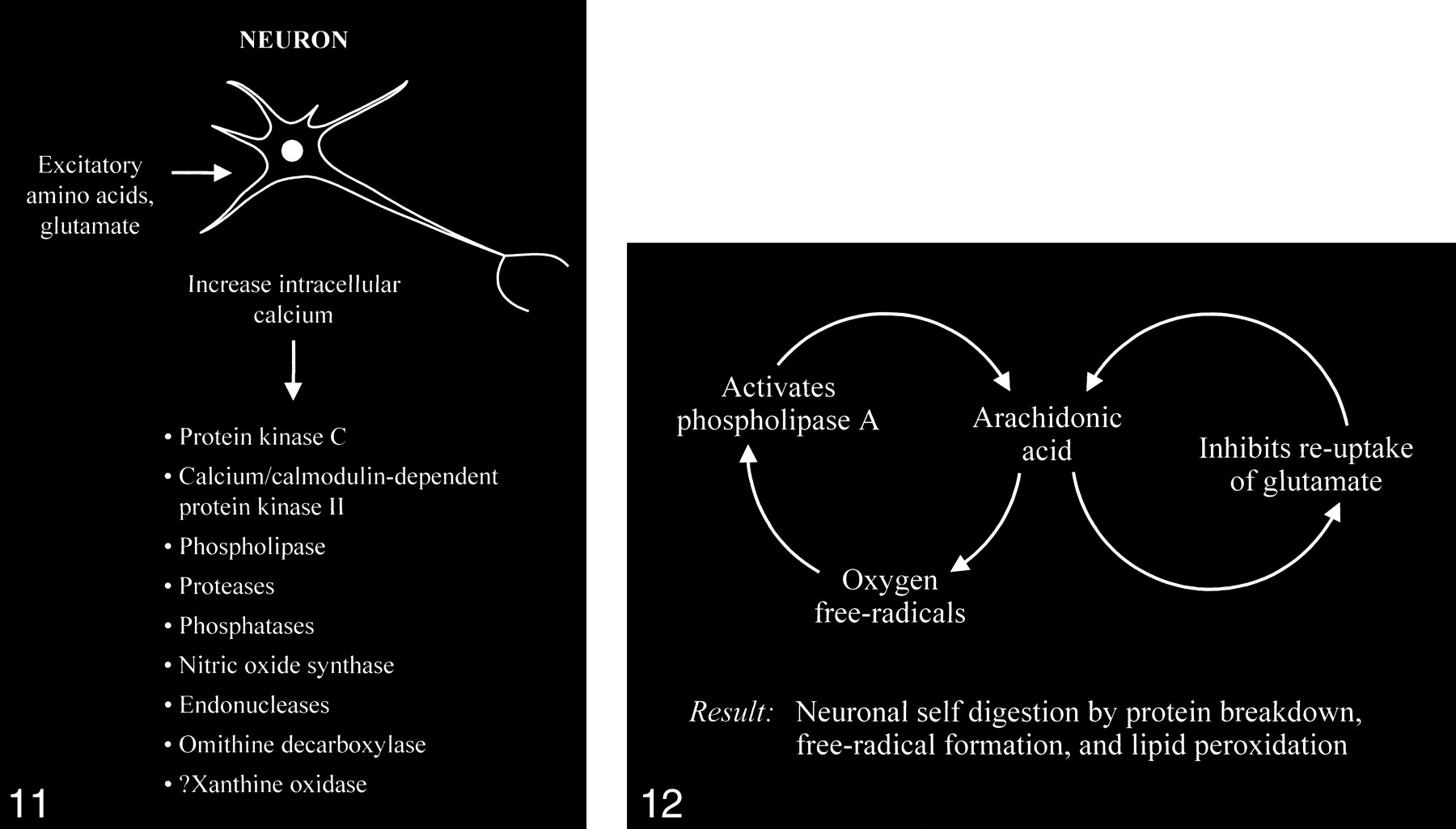
The accumulation of high intracellular calcium levels triggers a cascade of membrane, cytoplasmic, and nuclear events leading to neurotoxicity. Elevation of the intracellular calcium, however, appears to be a complex issue, because inducing similar intracellular calcium levels by using a metabolic inhibitor such as cyanide or membrane depolarization with potassium causes less permanent neuronal damage than with glutamate. The glutamate-induced elevated calcium levels proceed to overactivate a number of enzymes, including protein kinase C, calcium/cadmodulin-dependent protein kinase II, phospholipases, proteases, phosphatases, nitric oxide synthase, endonucleases, and ornithine decarboxylase (Fig 11). Some of these enzymes can also produce positive feedback loops to accelerate the downward spiral toward neuronal death (Fig 12). Activation of phospholipase A, for example, would generate platelet-activating factor and arachidonic acid and its metabolites. Platelet-activating factor directly contributes to the excitotoxic cascade by increasing glutamate release. Arachidonic acid inhibits reuptake of glutamate from the synaptic space, leading to further activation of glutamate receptors and more arachidonic acid formation. Increased arachidonic acid levels form oxygen free radicals, which activate phospholipase A, leading to more arachidonic acid formation. These enzymes and the generated feedback loops rapidly lead to neuronal self-digestion by protein breakdown, free radical formation, and lipid peroxidation.

Download figure / Download powerpoint
fig 11. Excessive accumulation of intracellular calcium caused by overactivation of the glutamate receptor sites stimulates multiple enzymes, which are involved in normal neuronal development and function, resulting in damage to the cell membrane, cytoskeleton, and DNA.
fig 12. Abnormally increased activation of some enzymes, such as phospholipase A, can cause an intracellular feedback cycle of events, leading to cell death.
Another important activated enzyme is nitric oxide synthase, which forms nitric oxide. Nitric oxide performs a variety of normal biological functions but the excessively stimulated NMDA receptors will produce abnormally increased levels of nitric oxide and superoxide ions. These substances may react and form peroxynitrite, which is extremely toxic, resulting in neuronal death. Nitric oxide can damage DNA as well as inhibit mitochondrial respiration, which in turn would create more free radicals and cause additional membrane depolarization. The nitric oxide–initiated neurotoxic cascades are important components of the mechanism of cell death in many neurodegenerative disorders, including Huntington disease (34–54).
Excessive Glutamate Accumulation
The key process that triggers the entire excitotoxic cascade is the excessive accumulation of glutamate in the synaptic space. This can be achieved by altering the normal cycling of intracranial glutamate (Fig 13) to increase the release of glutamate into the extracellular space or to decrease glutamate uptake/transport from the synaptic space, or by frank spillage of glutamate from injured neurons.

Download figure / Download powerpoint
fig 13. Neuronal glutamate that is released into the synaptic space is normally removed from the synaptic space by adjacent glial cells, in which the glutamate is converted to the closely related glutamine, which can then readily diffuse back into the neuron. Glutamine is converted back to glutamate in the neuron.
fig 14. Diagram shows sequence of events occurring in cerebral ischemia leading to neuronal death. (Free radical formation and lipase activation are also related to the increased intracellular calcium, although the two processes are not directly connected to the increased calcium by arrows in this schematic.)
Trauma is a blunt mechanism that massively elevates the extracellular glutamate levels. Normal extracellular glutamate concentration is about 0.6 μmol/L. Substantial neuronal excitotoxic injury occurs with glutamate concentrations of 2 to 5 μmol/L. Traumatic injury to neurons can produce disastrous results with the exposure of the normal intracellular glutamate concentrations of about 10 μmol/L to the extracellular space. Mechanical injury to a single neuron, therefore, puts all of the neighboring neurons at risk. Significant collateral injury occurs to surrounding neurons from this type of glutamate release. One recent therapeutic strategy is to immediately treat persons with injuries to the head or spinal column with glutamate receptor blockers to minimize the spread of neuronal death beyond the immediate physically disrupted neurons.
Several mechanisms of excess glutamate accumulation probably come into play in ischemia (Fig 14). Abnormal release of glutamate from its storage sites in neuronal vesicles is at least one factor. A feedback loop is generated as this released glutamate stimulates additional glutamate release. Ischemia also causes energy failure that impairs the reuptake by glutamate transporters. These transporters behave as symporters, which rely on the sodium gradient across cell membranes to move glutamate against its concentration gradients into the cell. The sodium gradient, however, is maintained by an energy-dependent pump that fails in ischemia. Such failure not only affects glutamate transport out of the synaptic space but also causes the transporters to run backward, becoming a source of extracellular glutamate rather than a sink for it. Ischemia deprives the neurons of oxygen and glucose, resulting in energy failure; however, energy failure itself is not particularly toxic to neurons. Neural toxicity occurs with the resultant activation of the cascade of glutamate receptor–dependent mechanisms. If these receptors are blocked by appropriate antagonists, the neurons can survive a period of deprivation of oxygen and metabolic substrate. This is the rationale for the recent development and trial of glutamate receptor blockers to treat acute ischemic events (55–66). While an infarcted zone cannot be salvaged, the hope is to prevent surrounding damage to the at-risk adjacent penumbra.
These receptor blockers may also be critical in the developing arena of interventional and pharmacologically related attempts to reestablish perfusion to acutely ischemic areas of the brain. Tissue reperfusion and increased oxygen concentrations to ischemic areas without concurrent halting of the excitotoxic cascade either at the receptor or intracellular levels may increase rather than decrease neuronal damage by providing additional free radicals in the form of superoxide anions as well as by increasing the intracellular cytosol calcium levels by stimulating the release of mitochondrial calcium stores.
Acceptance of the significant role of mitochondria in neuronal death and excitotoxicity is reflected in the rapidly expanding literature on this topic during the past decade (26, 49, 67–81). Preliminary investigations into the mechanisms of mitochondrial calcium homeostasis have already inspired several therapeutic neuroprotective strategies.
A number of drugs have been developed and used in an attempt to interrupt, influence, or temporarily halt the glutamate excitotoxic cascade toward neuronal injury (82–88). One strategy is the “upstream” attempt to decrease glutamate release. This category of drugs includes riluzole, lamotrigine, and lifarizine, which are sodium channel blockers. The commonly used nimodipine is a voltage-dependent channel (L-type) blocker. Attempts have also been made to affect the various sites of the coupled glutamate receptor itself. Some of these drugs include felbamate, ifenprodil, magnesium, memantine, and nitroglycerin. These “downstream” drugs attempt to influence such intracellular events as free radical formation, nitric oxide formation, proteolysis, endonuclease activity, and ICE-like protease formation (an important component in the process leading to programmed cell death, or apoptosis). Apoptosis occurs as part of the complex process of neuronal death, but many investigators believe that excitotoxicity and apoptosis are essentially different mechanisms that have intersecting influences (27, 56, 89–106).
Neuroradiologic Observations
Routine neuroimaging studies reflect the sequela of glutamate excitotoxic damage. The spreading involvement of adjacent brain tissue beyond the immediate area of insult in trauma and infarction is one example, and the delayed presentation of these conditions is another. The insidious brain parenchymal loss noted on imaging studies in patients with AIDS dementia is also thought to be related to glutamate excitotoxic injury.
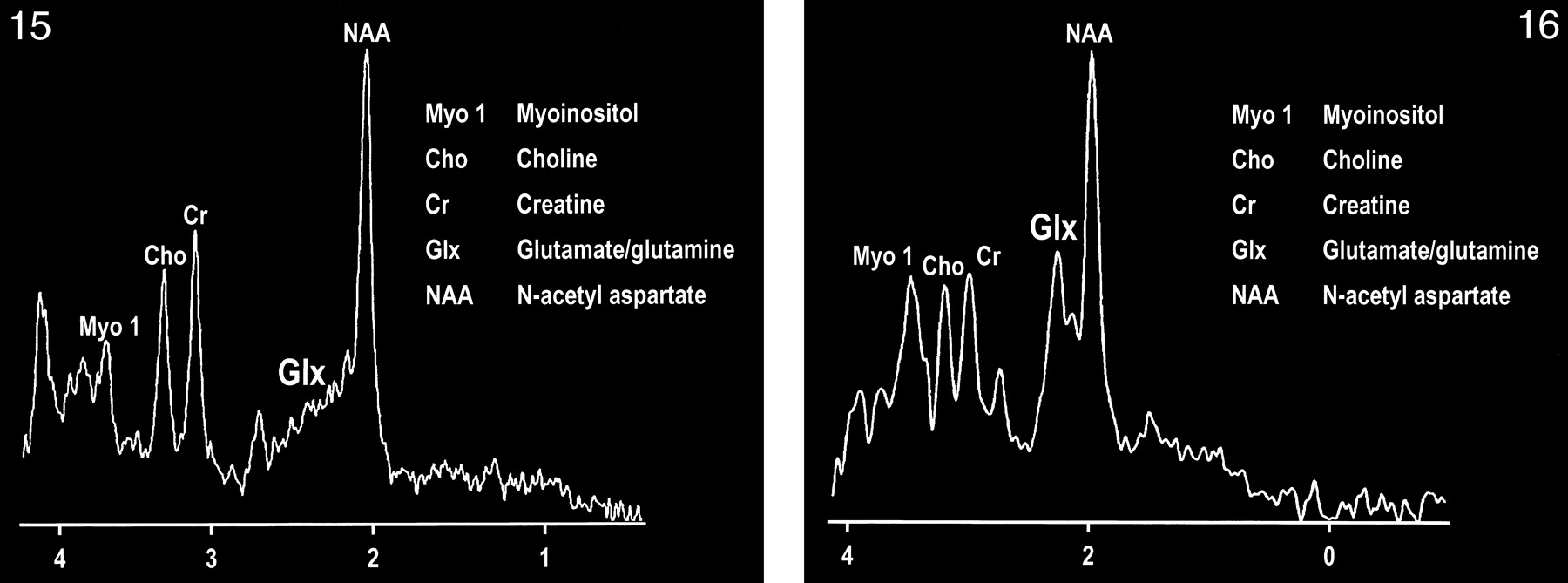
The development of spectroscopy at 0.5 T has provided an intriguing opportunity to observe the combined glutamate and glutamine (glx) peak in vivo (107). Delineation of this peak is enabled by the coalescence of the multiplets of glx at this field strength (Figs 15 and 16). The collapse of the γ and β multiplet resonances can be briefly summarized by the ratio of δ/J (δ = chemical shift, J = spin-spin coupling constant), which governs the spectral appearance of a strongly coupled multiplet structure such as glx. The spin-spin coupling constant is determined by molecular structure and is independent of field strength, whereas the chemical shift between coupled spins is a linear function of field strength. Therefore, as the ratio of δ/J approaches zero (or decreases), the multiplet collapses toward a single resonance line.
Download figure / Download powerpoint
fig 15. MR spectrum of a normal frontal lobe obtained at 1.5 T with a single-voxel point-resolved spectroscopy (PRESS) technique at 1500/41 (TR/TE).
fig 16. MR spectrum of the same frontal lobe as in figure 15 obtained at 0.5 T with a single-voxel PRESS technique at 1500/41. Note the more conspicuous glx peak.
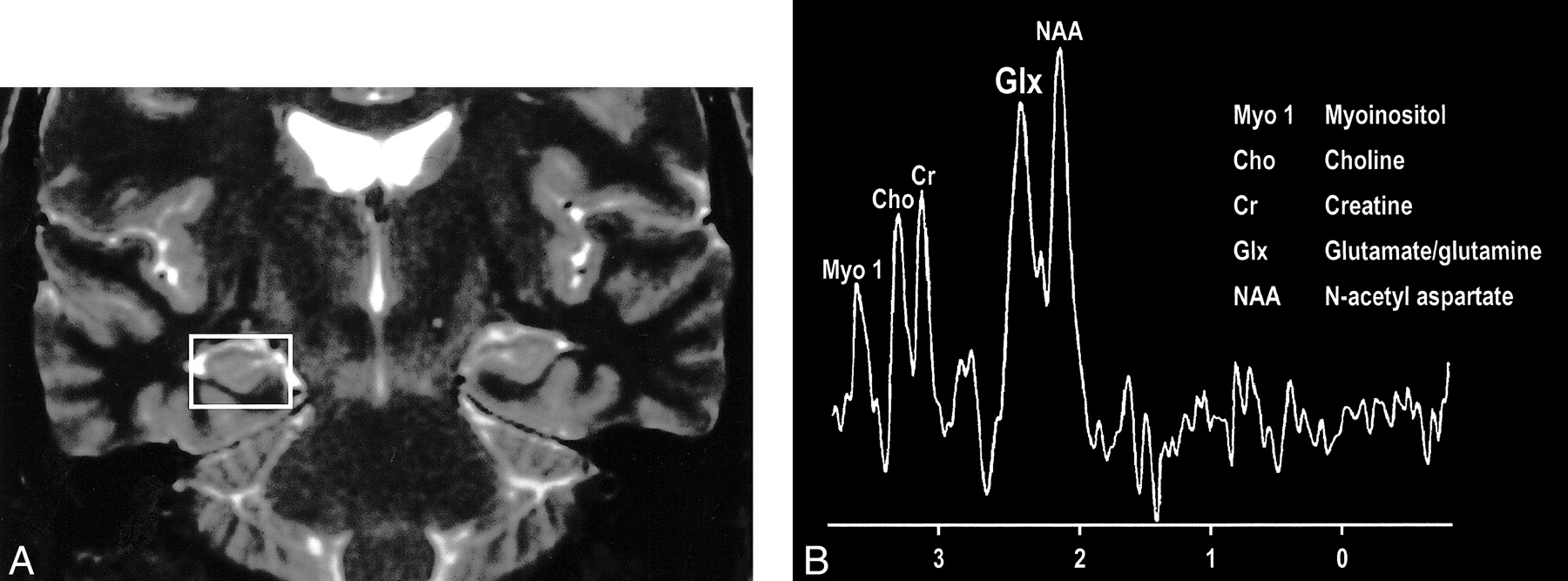
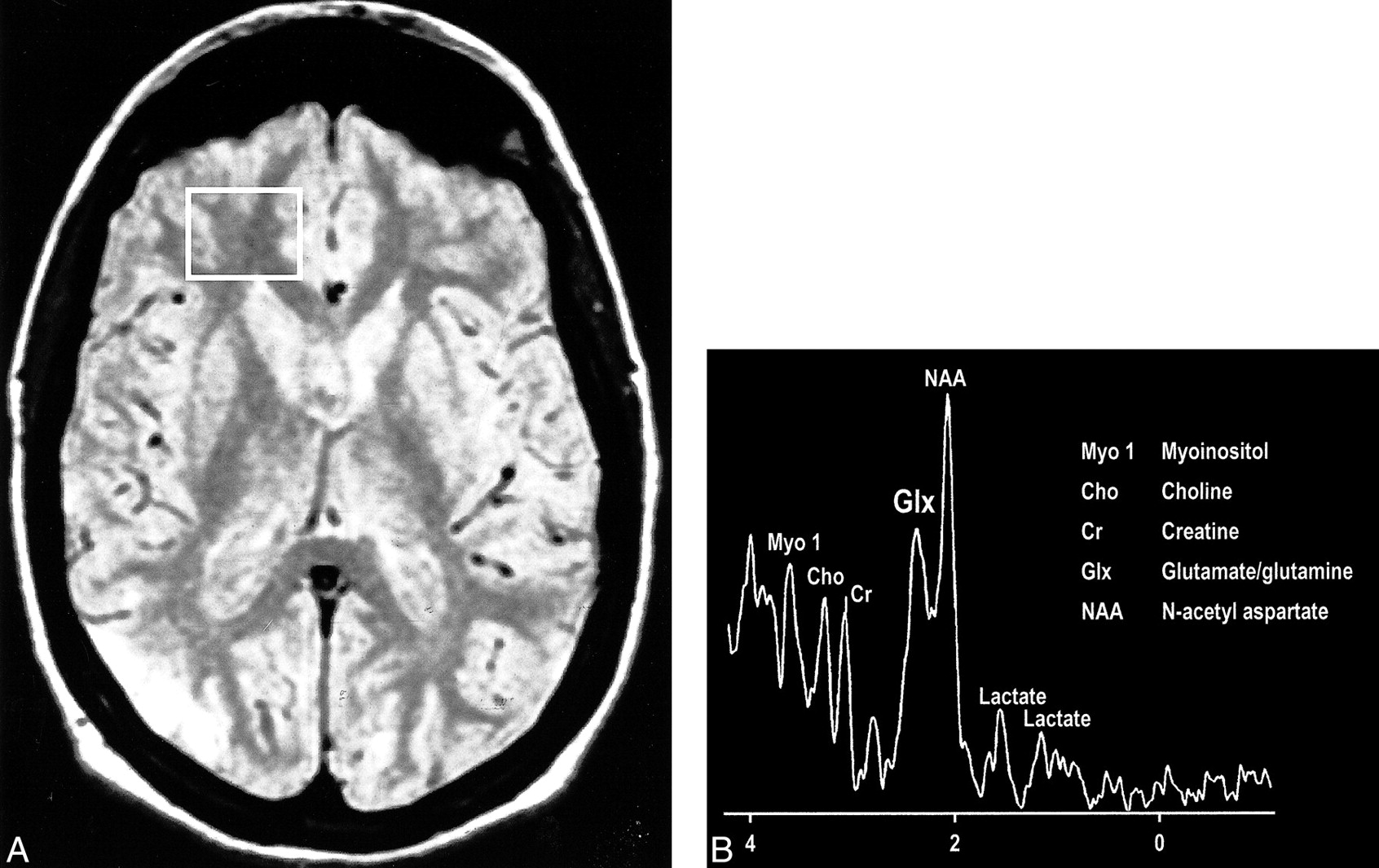
Interesting clinical observations are then possible and have been made with the ability to examine the glx peak. Glutamate excitotoxicity has been the implicated mechanism of neuronal injury in mesial temporal sclerosis with consistent, strong supportive experimental data (108–121). Elevated glx peaks have been observed in the hippocampi of these patients (Fig 17A and B) (122). Mitochondrial disorders, such as MELAS (mitochondrial myopathy, encephalopathy, lactacidosis, and stroke), are functionally ischemic, although there is no hypoperfusion but rather an inability to utilize the available oxygen. Imaging findings may be variable, but spectroscopic sampling of apparently normal-appearing areas of the brain by routine imaging can reveal not only the expected lactate peaks but also an elevated glx peak (Fig 18A and B). Even neurodegenerative disorders, such as Huntington disease, have been evaluated by this technique, and elevated glx peaks have been observed in the basal ganglia (123–125).

Download figure / Download powerpoint
fig 17. A, Coronal T2-weighted MR image of a patient with right mesial temporal sclerosis. B, MR spectrum obtained from a voxel (indicated in A) centered in the region of the patient’s right hippocampal formation shows an elevated glx peak.

Download figure / Download powerpoint
fig 18. Proton-density–weighted MR image in a patient with MELAS shows bilateral abnormal signal intensity changes at the periphery of the occipital lobes. B, MR spectrum obtained at 0.5 T shows elevated glx and lactate peaks from a sampling of the normal-appearing right frontal lobe of same patient (voxel placement indicated in A).
Summary
Glutamate excitotoxicity is the final common pathway resulting in neuronal injury for many seemingly unrelated disorders, including ischemia, trauma, seizures, hypoglycemia, hypoxia, and even some neural degenerative disorders. Familiarity with this process is important for neuroradiologists because of its central position in many of the disorders encountered in daily practice. This area has been one of the most intensely investigated fields in the neurosciences over the past several decades, and the information generated from this work will clearly influence our basic understanding of many neurologic disorders.”
Footnotes
-
1 Presented in part at the annual meeting of the American Society of Neuroradiology, San Diego, May 1999.
References
- Waggie KS, Kahle PJ, Tolwani RJ. Neurons and mechanisms of neuronal death in neurodegenerative diseases: a brief review. Lab Anim Sci 1999;49:358-362
- Olney J. Neurotoxicity of excitatory amino acids. In: McGeer E, Olney J, McGeer P, eds. Kainic Acid as a Tool in Neurobiology. New York: Raven Press; 1978:95–121
- Olney JW. Role of excitotoxins in developmental neuropathology. APMIS Suppl1993;40:103-112
- Budd SL. Mechanisms of neuronal damage in brain hypoxia/ischemia: focus on the role of mitochondrial calcium accumulation. Pharmacol Ther 1998;80:203-229
- Lipton SA, Nicotera P. Calcium, free radicals and excitotoxins in neuronal apoptosis. Cell Calcium 1998;23:165-171
- Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, eds. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. 5th ed. New York: Raven Press; 1994
- Aramburu J, Rao A, Klee CB. Calcineurin: from structure to function. Curr Top Cell Regul 2000;36:237-295
- Kader A, Frazzini VI, Solomon RA, Trifiletti RR. Nitric oxide production during focal cerebral ischemia in rats. Stroke 1993;24:1709-1716
- Choi D. Antagonizing excitotoxicity: a therapeutic strategy for stroke? Mount Sinai J Med 1998;65:133-138
- Gagliardi RJ. Neuroprotection, excitotoxicity and NMDA antagonists. Arq Neuropsiquiatr 2000;58:583-588
- Prost RW, Mark LP, Mewissen M, Li SJ. Detection of glutamate/glutamine resonances by 1H magnetic resonance spectroscopy at 0.5 tesla. Magn Reson Med 1997;37:615-618
- Sutula T, Koch J, Golarai G, Watanabe Y, McNamara JO. NMDA receptor dependence of kindling and mossy fiber sprouting: evidence that the NMDA receptor regulates patterning of hippocampal circuits in the adult brain. J Neurosci 1996;16:7398-7406
- Mark LP, Prost R, Yetkin Z, Haughton VM. An MR Spectroscopic Study of Glutamate in Patients with Temporal Lobe Seizures. Presented at the 33rd annual meeting of the American Society of Neuroradiology, Chicago, 1995
- Reynolds NC, Prost RW, Mark LP. Evidence for Excitotoxicity: Comparisons of Proton Magnetic Resonance Spectroscopy in Huntington’s Disease, Idiopathic Dystonia and Progressive Supranuclear Palsy. Presented at the 5th International Congress of Parkinson’s Disease and Movement Disorders, New York, 1998
- Received March 26, 2001, American Society of Neuroradiology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}